Coltura bidimensionale di MSC derivate dal cordone ombelicale
Le MSC del cordone ombelicale (ucMSC) sono state ottenute dalla Anthony Nolan Cord Bank, tagliate in piccoli pezzi e piastrate in terreno basale (ovvero, αMEM + 1% v/v penicillina-streptomicina) contenente il 5% (v/v) di piastrine umane lisato a 37 ° C, 5% CO2. Dopo la rimozione delle cellule non aderenti e il lavaggio con PBS, i terreni sono stati reintegrati e le cellule sono state coltivate fino a raggiungere una confluenza del 70–90%. È stata quindi eseguita la trypsinizzazione per un'ulteriore caratterizzazione e passaggio cellulare. Le ucMSC sono state caratterizzate per la positività di CD90, 105, CD106 e CD73, antigene leucocitario umano di classe I e per la mancanza di espressione di CD14, CD31 e CD45. Primi passaggi di 1×106 Le ucMSC (passaggi 2-5) sono state coltivate continuamente in un filtro filtrato da 175 cm2 fiasco (Corning) in terreno basale + 5% (v/v) di lisato piastrinico umano. Quando le cellule hanno raggiunto una confluenza del 70%-80%, i terreni sono stati sostituiti con terreno basale prima della raccolta del CCM il giorno successivo per l'isolamento dell'EV e la sottocoltura mediante un protocollo di trypsinizzazione convenzionale.
Cultura tridimensionale delle ucMSC
La piastra di coltura per micropozzetti Aggrewell400 è stata utilizzata per generare sferoidi ucMSC seguendo un protocollo precedentemente stabilito15. In breve, a ciascun pozzetto sono stati aggiunti 500 µl di soluzione di risciacquo anti-aderenza prima della centrifugazione a 2,000g per 2 minuti per rimuovere le bolle e incubare per 30 minuti–2 ore a temperatura ambiente. L'Aggrewell è stato quindi lavato con 500 µl di PBS per pozzetto, seguito dall'aggiunta di 500 µl di mezzo basale e dalla centrifugazione a 2,000g per 2 minuti per rimuovere le bolle. Il mezzo basale è stato poi sostituito con la sospensione cellulare ottenuta dalla trypsinizzazione 2D (preparata ad una densità di 1.2×105 cellule per 500 µl per pozzetto nel mezzo basale addizionato con il 20% v/v di siero sostitutivo KO secondo le istruzioni del produttore). La piastra è stata poi centrifugata a 200g per 5 minuti per l'aggregazione cellulare sul fondo di ciascun micropozzetto e mantenuti indisturbati nell'incubatore. Il CCM è stato raccolto il giorno 3 e ogni 2-3 giorni per l'isolamento dell'EV. Il mezzo fresco è stato reintegrato dopo la raccolta del mezzo per mantenere la coltura sferica di ucMSC per 12 giorni.
Isolamento dei veicoli elettrici
L'isolamento dell'EV è stato eseguito come descritto in dettaglio in precedenza46. Le provette da ultracentrifuga in policarbonato ultratrasparente (numero di catalogo 355631, Beckman Coulter) sono state riempite con 22.5 ml di CCM filtrato (preparato filtrando il CCM utilizzando un filtro per siringa da 0.22 µm). Quindi, 3 ml di soluzione di saccarosio al 25% p/p (preparata in D2O) è stato stratificato lentamente sotto il CCM utilizzando una pipetta Pasteur in vetro. La centrifugazione utilizzando un rotore oscillante (SW32 Ti, Beckman Coulter) è stata eseguita a 100,000g per 1.5 ore a 4 ° C. La soluzione di saccarosio è stata quindi raccolta (2 ml per provetta) e sottoposta a una fase di lavaggio per la purificazione dell'EV aggiungendo provette da ultracentrifuga in policarbonato preriempite (numero di catalogo 355618; Beckman Coulter) con 20 ml di PBS filtrato prima dell'ultracentrifugazione a 100,000g per 1.5 ore a 4 gradi centigradi utilizzando un rotore ad angolo fisso (70 Ti, Beckman Coulter). Il surnatante è stato scartato e il pellet di EV ottenuto è stato risospeso in PBS filtrato. Gli EV sono stati mantenuti a 4 °C per la conservazione di 1 settimana e a -80 °C per la conservazione a lungo termine.
Rilevazione dei marcatori EV mediante dot-blot
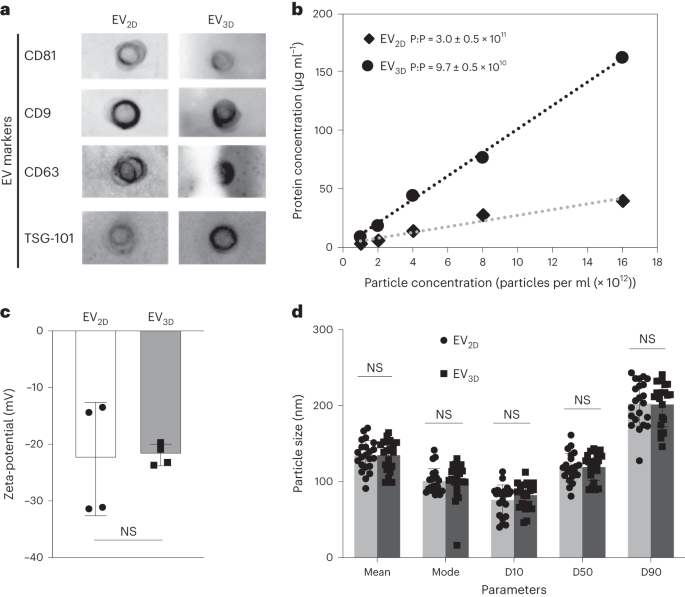
L'analisi è stata eseguita seguendo un protocollo precedentemente pubblicato15. Innanzitutto, 50 µl di EV ad una concentrazione di 5 × 1010 le particelle per ml sono state individuate su una membrana di nitrocellulosa (una membrana per marcatore) (Bio-Rad). La membrana è stata asciugata con azoto gassoso prima della fase di blocco utilizzando un tampone di blocco (ovvero latte scremato al 3% preparato in soluzione salina tamponata con Tris con Tween 0.1 allo 20% (TBS-T)) per 1 ora a temperatura ambiente. Gli anticorpi primari (CD9, CD63, CD81 e TSG101) sono stati aggiunti individualmente in tampone bloccante fresco seguito da incubazione per una notte a 4 °C (diluizione 1:1,000). La membrana è stata quindi lavata tre volte in TBS-T, 5 minuti per lavaggio. L'anticorpo secondario coniugato con perossidasi di rafano è stato quindi aggiunto al tampone bloccante fresco prima di un'ulteriore incubazione per 1 ora a temperatura ambiente (1:20,000 per anti-topo e 1:1,000 per anti-coniglio). La membrana è stata lavata come menzionato in precedenza e i segnali sono stati sviluppati mediante aggiunta di substrato (SuperSignal West Femto Maximum Sensitivity Substrate), seguita da imaging utilizzando il sistema Gel Doc (Bio-Rad) e analisi mediante il software Image Lab (Bio-Rad).
Saggio proteico per determinare la concentrazione proteica di veicoli elettrici isolati
Le concentrazioni proteiche sono state determinate mediante analisi microBCA in una piastra a 96 pozzetti seguendo le istruzioni del produttore adattate per i veicoli elettrici. In breve, i campioni EV (concentrazione minima, 5 × 1010 particelle per ml) sono state diluite 1:1 in PBS. La miscela di reagenti MicroBCA (preparata secondo le istruzioni del produttore) è stata aggiunta a duplicare 40 µl di campioni diluiti (50 µl per pozzetto), seguita dall'incubazione a 37 °C per 1 ora. La misurazione è stata confrontata con la BSA diluita serialmente come standard (preparata in duplicati). L'assorbanza è stata letta a 562 nm utilizzando un lettore di piastre FLUOStar Omega (BMG LabTech). Per l'analisi è stato utilizzato il software MARS v.2.40 (BMG LabTech) estrapolando i valori dalla curva standard utilizzando un'equazione polinomiale di terzo ordine, con r2 > 0.999 per ciascun test.
NTA
La dimensione e la concentrazione dei veicoli elettrici sono state misurate da NTA utilizzando un Nanosight LM10 (Malvern Instruments) dotato di un laser da 488 nm. Il livello della fotocamera è stato regolato automaticamente e la soglia di rilevamento dell'analisi è stata impostata su 3–4. I campioni di EV sono stati diluiti in PBS filtrato per ottenere concentrazioni ottimali (20–80 particelle per fotogramma). Per ciascuna preparazione del veicolo elettrico sono state effettuate quattro registrazioni video della durata di 40 s. Per analizzare il video registrato è stato utilizzato il software Nanosight NTA 3.2 (Malvern Instruments).
Potenziale Zeta
La mobilità elettroforetica dinamica di EV e HC-EV è stata misurata con un software Malvern Zetasizer Nano ZS e Zetasizer v.7.12 (Malvern Instruments). Prima delle misurazioni, i campioni di EV (concentrazione minima, 1 × 1011 particelle per ml) sono state diluite 1:50 in acqua deionizzata. Le misurazioni sono state effettuate a 25 °C per ciascun triplicato sperimentale.
Preparazione della corona proteica
Il rivestimento della corona proteica è stato eseguito seguendo un protocollo pubblicato47 con modifiche. Per rimuovere gli EV e il siero proteico aggregato, FBS è stato sottoposto a ultracentrifugazione a 100,000g per 18 ore a 4 °C. Il surnatante è stato raccolto e filtrato attraverso filtri da 0.22 µm (EV-D FBS). Veicoli elettrici (~7 × 1011 particelle, equivalenti a 0.05 m2) sono stati preparati in un volume totale di 300 µl di PBS sterile integrato con una soluzione di penicillina-streptomicina all'1% (sPBS-PS) in provette Eppendorf da 1.5 ml prima dell'incubazione con 300 µl di EV-D FBS per 1 ora a 37 °C, 300 giri/min. Dopo l'incubazione, la miscela è stata trasferita in una provetta da ultracentrifuga (numero di catalogo 343778, Beckman Coulter). I campioni sono stati ultracentrifugati a 100,000g per 1 ora a 4 °C. Il surnatante è stato accuratamente rimosso senza disturbare il pellet. Il pellet è stato ulteriormente lavato con 1 ml di sPBS-PS due volte, seguendo le condizioni sopra menzionate. Negli studi sul desorbimento corona, dopo il lavaggio finale, il pellet (HC-EV) è stato disperso in 100 µl di SDS al 2% p/v appena preparato, soluzione di Tris-HCl 62.5 mM, seguita da incubazione a 95 °C per 5 minuti per desorbire le proteine che costituiscono l'HC. L'HC è stato quindi separato dagli EV mediante ultracentrifugazione come sopra. Il surnatante contenente HC è stato raccolto e sottoposto a determinazione proteica mediante saggio microBCA. Le soluzioni HC sono state conservate a -80 °C prima di ulteriori analisi.
SDS–PAGINA
I campioni EV, HC-EV, EV-D FBS, KO e HC (15 µg) sono stati miscelati con tampone campione LDS e tampone RIPA (miscelato con cocktail di inibitori della proteasi 1:100) e incubati per 10 minuti a 70 °C. I campioni sono stati quindi applicati su un gel proteico NuPAGE 4–12% Bis–Tris e fatti funzionare per 30 minuti a 100 mV e un'ulteriore 1 ora a 150 mV. Per la colorazione con argento, il gel è stato fissato in 100 ml di soluzione di fissaggio (50% metanolo, 10% acido acetico, 50 µl di formaldeide) durante la notte, seguito da lavaggio con etanolo al 50% tre volte. Il gel è stato quindi sensibilizzato mediante ipo-soluzione (0.02% p/v di tiosolfato di sodio) per 1 minuto, seguita dall'impregnazione del gel con 0.2% p/v di nitrato d'argento per 30 minuti. Il gel è stato quindi lavato con acqua deionizzata tre volte e sottoposto a sviluppo di bande utilizzando 100 ml di soluzione di sviluppo (6 g di carbonato di sodio, 2 ml di iposoluzione e 50 µl di formaldeide). Una volta sviluppata, la reazione è stata fermata con acido acetico al 5% v/v. È stata scattata una fotografia digitale del gel su uno sfondo bianco.
Digestione della corona proteica per analisi di spettrometria di massa
L'analisi proteomica delle proteine FBS, degli EV e dei loro HC mediante LC-MS è stata condotta utilizzando il metodo descritto da Schottler et al.47. In breve, l'SDS è stato rimosso dai campioni mediante colonne di rimozione del detergente Pierce e 25 µg di ciascun campione proteico sono stati precipitati mediante il kit di precipitazione proteica ProteoExtract seguendo il manuale del produttore. I pellet proteici risultanti sono stati risospesi in RapiGest SF allo 0.1% in bicarbonato di ammonio 50 mM e incubati per 15 minuti a 80 °C. Ditiotreitolo (concentrazione finale, 5 mM) è stato aggiunto per ridurre le proteine e la miscela è stata incubata a 56°C per 45 minuti. È stata quindi aggiunta iodoacetammide (concentrazione finale, 15 mM) e la miscela è stata ulteriormente incubata al buio per 1 ora a temperatura ambiente. I campioni proteici sono stati digeriti per 18 ore a 37 °C mediante trypsin con un rapporto enzima:proteina di 1:50 (p/p). La reazione è stata interrotta mediante l'aggiunta di 2 μl di acido cloridrico. I peptidi digeriti sono stati diluiti con acido formico allo 0.1% v/v in acqua di grado UPLC-MS e addizionati con 50 fmol μl-1 Hi3 Escherichia coli standard per la quantificazione assoluta.
Assorbimento cellulare in vitro di EV e HC-EV
EV2D ed EV3D sono stati etichettati secondo il nostro protocollo pubblicato48. La concentrazione in particelle per ml è stata ottenuta mediante NTA come menzionato sopra. L'intensità della fluorescenza degli EV appena preparati (100 µl) è stata misurata utilizzando un lettore di piastre FLUOStar Optima (BMG Labtech), con lunghezze d'onda di eccitazione ed emissione di 485 nm e 520 nm, rispettivamente. Quando c'è una differenza nell'efficienza di etichettatura tra i lotti o i tipi di EV, i campioni sono stati miscelati con EV non etichettati in modo che l'intensità della fluorescenza e il numero di particelle fossero comparabili per i diversi campioni. Le cellule fagocitiche, cioè J774 e macrofagi derivati da monociti umani, e le cellule non fagocitiche, cioè HepG2, sono state seminate ad una densità di 1.5 × 105 cellule per pozzetto in piastre da 24 pozzetti (Corning). Dopo 24 ore, i mezzi sono stati cambiati in mezzi privi di siero e le cellule sono state trattate con EV marcato2D, HC-EV2D, EV3D e HC-EV3D di valori di unità di fluorescenza comparabili e una dose di 2 × 109 particelle e incubate per 1 ora, 4 ore e 24 ore. Per valutare il contributo del recettore legante l'albumina nel mediare l'internalizzazione dell'EV, le cellule HepG2 sono state trattate con BSA (12.5 mg ml-1), seguito dall'aggiunta di EV marcati con DiD3D, per 24 ore. La citometria a flusso è stata eseguita su un BD FACSCalibur utilizzando il software BD FACStation v.6.0 (BD Biosciences) sulle cellule staccate mediante trypsin–EDTA 0.05%, lavate e risospese in 200 µl di PBS. Tutti i campioni sono stati analizzati utilizzando FlowJo v.10.7.2 (TreeStar/BD Bioscience). Le cellule erano delimitate dalla loro dispersione anteriore e laterale. L'assorbimento di EV è stato valutato dall'MFI di Alexa Fluor 488. La variazione di piega nell'MFI è stata calcolata rispetto all'aumento relativo dell'MFI rispetto ai controlli non trattati.
Animali
Tutti gli esperimenti sugli animali sono stati eseguiti in conformità con la legge britannica sugli animali (procedure scientifiche) del 1986 e con il codice di condotta del Ministero degli interni del Regno Unito per l'alloggiamento e la cura degli animali utilizzati nelle procedure scientifiche (Ministero degli Interni 1989). La sperimentazione in vivo ha aderito alla licenza del progetto approvata dall'organismo di revisione etica e per il benessere degli animali del King's College di Londra (AWERB) e dal Ministero degli Interni del Regno Unito (PBE6EB195 e PP8950634). La ricerca sugli animali e le cure veterinarie sono state eseguite presso il Franklin-Wilkins Building, King's College di Londra, secondo il protocollo approvato per questo studio da un responsabile della formazione e delle competenze designate (Julie Keeble) e da un responsabile della cura e del benessere degli animali (Jayne Morgan). Topi CD-1 femmine (~ 25-35 g, 5 settimane di età) sono stati ottenuti da Charles River per lo studio sulla biodistribuzione in vivo. Topi C57BL/6 maschi e femmine (18–25 g, 6–8 settimane di età) sono stati ottenuti da Charles River per lo studio sull'assorbimento cellulare in vivo. Tutti i topi sono stati alloggiati in un ciclo di 12 ore di luce/12 ore di buio con la temperatura mantenuta tra 65 e 75 °F (~18–23 °C) e ~50% di umidità. Nello studio sono stati utilizzati entrambi i sessi, in conformità con le raccomandazioni recentemente pubblicate dal Medical Research Council del Regno Unito per la conduzione di ricerche sugli animali.
Biodistribuzione in vivo di veicoli elettrici mediante imaging ottico a fluorescenza
Il CCM filtrato è stato incubato con 1 µM di tracciante lipofilo fluorescente DiR (1,1-diottadecil-3,3,3,3-tetrametilindotricarbocianina ioduro) a temperatura ambiente per 1 ora con agitazione prima dell'isolamento dell'EV mediante cuscino di saccarosio più ultracentrifugazione come descritto sopra per rimuovere il colorante non legato. L'esperimento è stato eseguito su topi CD1, divisi casualmente in gruppi. EV marcato DiR appena purificato2D ed EV3D (2×1011 particelle in 200 µl) sono state iniettate per via endovenosa attraverso la vena della coda. Ai topi di controllo è stato iniettato solo PBS o PBS contenente DiR. Topi vivi sedati con isoflurano sono stati sottoposti a imaging utilizzando il sistema IVIS Lumina III (eccitazione, 740 nm; emissione, 840 nm) a 1, 4 e 24 ore dopo l'iniezione endovenosa prima di uccidere gli animali. Gli organi principali (cervello, cuore, polmone, fegato, stomaco, milza, rene e intestino) sono stati raccolti per l'imaging a fluorescenza ex vivo. L'intensità della fluorescenza in ciascun organo (efficienza radiante totale) è stata ottenuta utilizzando il software Living Image v.4.7.3 (PerkinElmer) per determinare la biodistribuzione negli organi di EV marcati con DiR2D ed EV3D disegnando le regioni di interesse (ROI) di ciascun organo. I valori sono stati poi normalizzati al peso degli organi (efficienza radiante totale per g). Per lo studio sulla clearance renale, dopo l'iniezione con EV marcati con DiR, i topi sono stati alloggiati individualmente in una gabbia metabolica circolare standard (Nalgene Nunc) per 24 ore e l'urina è stata raccolta in una provetta Nalgene sul fondo di un sistema a imbuto per ulteriore fluorescenza determinazione dell'intensità utilizzando un sistema IVIS Lumina III e il software Living Image v.4.7.3 (PerkinElmer) disegnando ROI che coprono un contenitore di urina per ottenere l'efficienza radiante totale per campione di urina.
Assorbimento cellulare in vivo di EV nelle sottopopolazioni epatiche
EV2D ed EV3D sono stati etichettati con DiD (DiIC18(5); 1,1′-diottadecil-3,3,3′,3′-tetrametilindodicarbocianina, sale 4-cloro-benzensolfonato) utilizzando la stessa procedura utilizzata per l'etichettatura DiR negli studi in vivo. L'esperimento è stato eseguito su topi C57BL/6, divisi casualmente in gruppi. EV marcato con DiD appena purificato2D ed EV3D (2×1011 particelle in 200 µl) sono state iniettate per via endovenosa attraverso la vena della coda. Ai topi di controllo è stato iniettato solo PBS o PBS contenente DiD. Per lo studio sul blocco dei recettori dell'albumina, 100 µl di BSA diluiti in PBS (10 mg ml-1, dosaggio selezionato in base alla letteratura49,50,51,52) è stato iniettato 5 minuti prima dell'EV3D iniezione (2 × 1011 particelle in 100 µl). Per lo studio dell'internalizzazione di EV rivestiti con albumina, per preparare EV rivestiti con albumina e marcati con DiD2D, ucMSC 2D (confluenza del 70–80%) sono state coltivate in terreno basale integrato con 2.5 mg ml-1 BSA, seguita dalla raccolta del terreno dopo 24 ore di coltura, etichettatura DiD e isolamento EV come menzionato in precedenza. EV marcato con DiD rivestito di albumina2D sono stati poi iniettati per via endovenosa (2 × 1011 particelle in 200 µl). Ventiquattro ore dopo, i topi sono stati anestetizzati con fenobarbital e sottoposti a dissezione cutanea sulla linea mediana ventrale per aprire la cavità peritoneale per la perfusione epatica seguendo il protocollo precedentemente pubblicato con modifiche53. I topi sono stati perfusi con 30 ml di HBSS–EGTA riscaldato a 41 °C attraverso la vena cava inferiore utilizzando una pompa peristaltica (SciQ 300, Watson Marlow) e un set di infusione alato 27G × 0.38″ × 12″ (BD Valu-Set) ad una velocità di 15 giri al minuto La vena porta epatica è stata tagliata dopo che il fegato si è gonfiato e scolorito, dopo di che la velocità è stata regolata a 20 giri al minuto. I topi sono stati quindi perfusi con 25 ml di HBSS–CaCl contenente collagenasi2 (concentrazione, 1 mg ml-1) riscaldato a 41 °C, a una velocità di 15 giri/min. Il fegato è stato trasferito in una capsula Petri contenente HBSS–CaCl freddo2 respingente. Le cellule epatiche sono state rilasciate rompendo delicatamente la capsula di Glisson e agitando il fegato. Una volta ottenuta la sospensione cellulare omogenea, le cellule sono state filtrate attraverso un colino da 70 µm (Corning). Gli epatociti sono stati separati mediante centrifugazione per 3 minuti a 50g a 4 °C con freno basso, tre giri. Dopo ogni giro, il pellet è stato risospeso in 30 ml di HBSS–CaCl freddo2e il surnatante è stato raccolto per un ulteriore frazionamento NPC. La frazione NPC è stata pellettizzata a 650g a 4 °C e sottoposto ad incubazione con tampone di lisi RBC per 5 minuti su ghiaccio, seguito dall'aggiunta di 20 ml di PBS per arrestare la reazione e ulteriore centrifugazione a 650g a 4 ° C per far sedimentare la frazione NPC purificata. L'identificazione di ciascuna sottopopolazione è stata eseguita in base all'espressione del marcatore utilizzando anticorpi marcati con fluorescenza per l'analisi mediante citometria a flusso (BD FACSCelesta, gestito dal software BD FACSDiva v.9.2, BD Biosciences) e dal software FlowJo v.10.7.2 (TreeStar/BD Bioscience ). Gli epatociti sono stati colorati con anticorpo anti-topo ASGPR1 (1:200)54,55,56 e anti-CD9 umano tramite colorazione intracellulare utilizzando Triton-X 0.1 allo 100% in PBS. Le cellule di Kupffer sono state colorate con anti-topo CD45, F4-80 e CD11b (1:200, ciascuno)57,58,59. Le cellule endoteliali sono state colorate con anti-topo CD45 (1:200), CD31 (1:200) e CD146 (1:100)60,61,62. Le cellule stellate sono state colorate con anti-topo CD45 (1:200), GFAP (1:50) e rilevate utilizzando un filtro 450/50 e un laser viola da 405 nm63,64,65. Tutte le frazioni sono state colorate con Zombie Aqua per determinare la vitalità delle cellule. La colorazione è stata eseguita per 30 minuti a temperatura ambiente. La frequenza delle cellule madri che assumono EV è stata definita separando la popolazione di cellule positive al DiD rispetto al gruppo di controllo (PBS). Gli MFI del segnale DiD espresso da ciascun tipo di cellula sono stati utilizzati per valutare la quantità di assorbimento degli EV.
analisi statistica
Le analisi statistiche dei dati sono state eseguite utilizzando Prism 9.4.1 (software GraphPad) utilizzando l'analisi della varianza a una via (ANOVA) con il test post hoc di Tukey per tutti P valori (*P < 0.05, **P <0.01, ***P < 0.001, ****P <0.0001; P > 0.05 non era significativo). Tutti i risultati sono espressi come media ± DS Si presume che la distribuzione dei dati sia normale ma ciò non è stato formalmente testato. Non sono stati utilizzati metodi statistici per predeterminare le dimensioni del campione, ma le dimensioni del campione sono state scelte sulla base di precedenti esperimenti eseguiti in modo simile dal nostro gruppo con effetti statisticamente significativi comprovati10. Tutti i grafici sono stati realizzati in Prism 9.4.1. MATLAB 9.11 è stato utilizzato per generare mappe di calore dei dati. La raccolta e l'analisi dei dati non sono state eseguite in modo cieco rispetto alle condizioni degli esperimenti. Tutti i punti dati sono stati inclusi per le analisi.
Reporting summary
Ulteriori informazioni sulla progettazione della ricerca sono disponibili nel Riepilogo dei rapporti sul portafoglio naturalistico collegato a questo articolo.
- Distribuzione di contenuti basati su SEO e PR. Ricevi amplificazione oggi.
- PlatoData.Network Generativo verticale Ai. Potenzia te stesso. Accedi qui.
- PlatoAiStream. Intelligenza Web3. Conoscenza amplificata. Accedi qui.
- PlatoneESG. Carbonio, Tecnologia pulita, Energia, Ambiente, Solare, Gestione dei rifiuti. Accedi qui.
- Platone Salute. Intelligence sulle biotecnologie e sulle sperimentazioni cliniche. Accedi qui.
- Fonte: https://www.nature.com/articles/s41565-023-01585-y
- :È
- :non
- ][P
- $ SU
- 000
- 001
- 01
- 05
- 1
- 10
- 100
- 11
- 12
- 15%
- 150
- 16
- 175
- 1996
- 2%
- 20
- 200
- 2000
- 2010
- 2011
- 2013
- 2015
- 2016
- 2017
- 2018
- 2019
- 2020
- 2021
- 22
- 24
- 25
- 2D
- 30
- 300
- 35%
- 4
- 40
- 41
- 45
- 46
- 48
- 5
- 50
- 500
- 52
- 54
- 58
- 6
- 62
- 65
- 7
- 70
- 75
- 80
- 89
- 9
- a
- sopra
- Assoluta
- Secondo
- con precisione
- acquisire
- Legge
- adattato
- aggiunto
- l'aggiunta di
- aggiunta
- aderito
- Rettificato
- amministrato
- Dopo shavasana, sedersi in silenzio; saluti;
- contro
- aggregato
- aggregazione
- AL
- Alexa
- Tutti
- am
- quantità
- an
- analizzare
- analisi
- .
- Presentatrice
- ed
- animali
- animali
- Anthony
- Anticorpi
- anticorpo
- antigene
- applicato
- approvato
- acqua
- SONO
- articolo
- AS
- assunto
- At
- automaticamente
- disponibile
- b
- sfondo
- BAND
- Banca
- basato
- BD
- BE
- è diventato
- prima
- sotto
- fra
- biomarcatori
- cieco
- blocco
- stile di vita
- entrambi
- Parte inferiore
- Cervello
- Rottura
- brevemente
- BSA
- bufferizzare
- Costruzione
- ma
- by
- calcolato
- stanza
- Cancro
- Cellule cancerogene
- capace
- Ultra-Grande
- che
- attentamente
- svolta
- cella
- Celle
- cellulare
- il cambiamento
- cambiato
- caratterizzato
- Charles
- scelto
- circolare
- classe
- autorizzazione
- clicca
- cocktail
- codice
- freddo
- raccolto
- collezione
- College
- colonne
- paragonabile
- rispetto
- conformità
- conforme
- concentrazione
- condizione
- condizioni
- condotto
- conduzione
- Connolly
- Contenitore
- continuamente
- contributo
- di controllo
- controlli
- convenzionale
- cordone
- Corona
- Consiglio
- copertura
- Cultura
- curva
- taglio
- ciclo
- Scuro
- dati
- punti dati
- giorno
- Giorni
- de
- definito
- consegna
- densità
- dipendente
- descritta
- Design
- dettaglio
- rilevato
- rivelazione
- determinazione
- Determinare
- determinato
- determinazione
- sviluppato
- in via di sviluppo
- Mercato
- DID
- differenza
- diverso
- digitale
- diluito
- diluizione
- scartato
- piatto
- dispersi
- distinto
- distribuzione
- Diviso
- DOE
- dosaggio
- dose
- disegno
- duplicati
- durata
- durante
- dinamico
- disfunzione
- e
- E&T
- ogni
- Presto
- effetto
- effetti
- efficienze
- efficienza
- emissione
- abilitato
- migliorata
- Migliora
- equazione
- attrezzato
- Equivalente
- sviluppate
- Etere (ETH)
- etico
- EV
- valutare
- valutato
- valutazione
- Ogni
- evs
- esperimento
- sperimentale
- esperimenti
- espresso
- espressione
- la donna
- pieno
- filtro
- filtraggio
- filtri
- finale
- Nome
- fisso
- flusso
- seguito
- i seguenti
- Nel
- formaldeide
- formalmente
- formazione
- Avanti
- quattro
- frazione
- TELAIO
- Frequenza
- fresco
- da
- function
- funzionalmente
- ulteriormente
- GAS
- gated
- cancello
- generare
- gentile
- vetro.
- grado
- grafici
- Gruppo
- Gruppo
- Raccolta
- Cuore
- Casa
- Home Office
- ORE
- alloggiamento
- HTTPS
- umano
- i
- ICE
- Identificazione
- identifica
- iii
- Immagine
- Imaging
- immune
- immunità
- immunomodulante
- in
- incluso
- Aumento
- incubato
- Incubando
- INCUBAZIONE
- incubatrice
- Individualmente
- induce
- infiammazione
- infiammatorio
- informazioni
- infusione
- istruzioni
- strumenti
- interazione
- interesse
- ai miglioramenti
- endovenoso
- per via endovenosa
- isolato
- da solo
- tenere
- rene
- uccisione
- kit
- laboratorio
- etichettatura
- Dipingere
- carente
- laser
- dopo
- stratificato
- Livello
- Licenza
- limiti
- LINK
- connesso
- vivere
- Fegato
- vita
- Londra
- a lungo termine
- Basso
- luce
- macrofagi
- fatto
- mantenere
- mantenuto
- maggiore
- maschio
- Manuale
- marcatore
- marzo
- Massa
- materiale
- massimo
- significare
- misurato
- misurazioni
- Media
- medicale
- ricerca medica
- medie
- menzionato
- metabolica
- Metanolo
- metodo
- metodi
- metodi erano
- topi
- migrazione
- latte
- verbale
- ordine
- minore
- scelta
- misto
- miscela
- ML
- mobilità
- modifiche
- MOL
- Morgan
- mouse
- Detto
- nano
- nanotecnologia
- Natura
- no
- normale
- numero
- numeri
- ottenere
- ottenuto
- ottenendo
- of
- Office
- Responsabile
- Vecchio
- on
- una volta
- ONE
- esclusivamente
- aprire
- operato
- ottico
- ottimale
- or
- nostro
- su
- ancora
- per una notte
- passaggi
- PBS
- per
- eseguita
- Petri
- fenotipo
- pezzi
- forare
- Platone
- Platone Data Intelligence
- PlatoneDati
- più
- punti
- polinomio
- popolazione
- Portale
- lavori
- Positività
- Post
- pratica
- preparazione
- Preparare
- preparato
- precedente
- in precedenza
- primario
- Precedente
- procedura
- procedure
- progetto
- proliferazione
- promuoverlo
- protegge
- protettivo
- Proteine
- Proteine
- protocollo
- comprovata
- pubblicato
- pompa
- quantificazione
- R
- Radiante
- rapporto
- rbc
- a raggiunto
- raggiungendo
- reazione
- Leggi
- Lettore
- recentemente
- ricevitore
- raccomandazioni
- registrato
- ridurre
- riferimento
- riflettere
- regioni
- normativo
- parente
- rilasciato
- rimozione
- rimuovere
- rimosso
- renale
- sostituito
- sostituzione
- Reportistica
- necessario
- riparazioni
- rispetto
- risultante
- Risultati
- ritornando
- recensioni
- fiume
- Prenotazione sale
- Correre
- s
- sale
- stesso
- campione
- SCI
- scientifico
- secondario
- selezionato
- prodotti
- Sensibilità
- Sepsi
- Siero
- set
- lato
- Signal
- Segnali
- significativa
- Argento
- Allo stesso modo
- singolo
- Taglia
- Dimensioni
- scremare
- Pelle
- Lentamente
- piccole
- So
- sodio
- Software
- soluzione
- Soluzioni
- velocità
- Spin
- giri
- Standard
- statistiche
- statisticamente
- Stealth
- gambo
- cellule staminali
- step
- Fermare
- fermato
- conservazione
- memorizzati
- studi
- Studio
- substrato
- superficie
- sospensione
- sistema
- T
- preso
- presa
- mira
- test
- testato
- che
- I
- Regno Unito
- loro
- poi
- Là.
- questo
- tre
- tridimensionale
- soglia
- Attraverso
- volte
- a
- Totale
- Tracer
- Training
- trasferito
- trattati
- Due volte
- seconda
- Digitare
- Tipi di
- Uk
- per
- unità
- fino a quando
- su
- comprensione
- utilizzato
- utilizzando
- Valori
- veterinario
- via
- vitalità
- Video
- vivo
- volume
- Prima
- lavaggio
- Water
- Watson
- lunghezze d'onda
- settimana
- Settimane
- Benessere
- WELL
- sono stati
- ovest
- Wheeler
- quando
- quale
- bianca
- con
- entro
- senza
- i rendimenti
- zefiro
- zombie