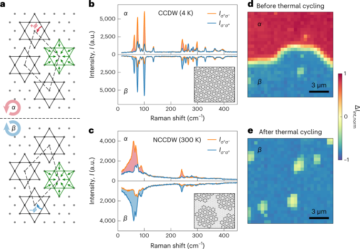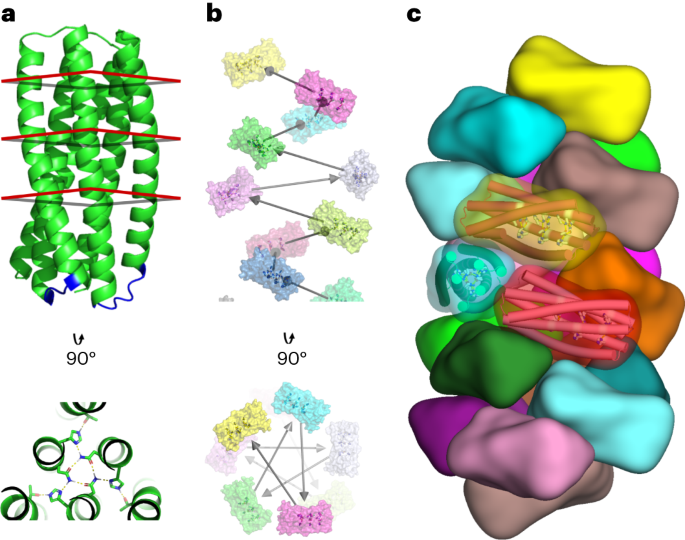
Strategia projektowania obliczeniowego
Krótkie pętle do łączenia helis pro-2.3 w pojedynczy łańcuch zaprojektowano przy użyciu wyczerpującej bazy danych próbek szkieletu składających się z fragmentów obejmujących dwa obszary helikalne zidentyfikowane za pomocą DSSP w strukturach krystalograficznych o wysokiej rozdzielczości (jak opisano wcześniej14). W tej bazie danych zidentyfikowano pętle poprzez sztywne dopasowanie reszt końcowych fragmentu i celu, stosując zoptymalizowany algorytm superpozycji15. Kandydaci, którzy spełnili tolerancję wyrównania wynoszącą 0.35, XNUMX Å RMSD, zostali dopasowani do docelowego szkieletu za pomocą współrzędnych przestrzeni skrętnej i miękkich ograniczeń współrzędnych do dopasowanych współrzędnych ciężkich atomów szkieletu kandydata. Następnie zaprojektowano sekwencje pętli kandydujących w oparciu o ograniczenia profilu sekwencji generowane przez dopasowanie szkieletu pętli do bazy danych struktury źródłowej. Do ostatecznego projektu pętli zostali wybrani kandydaci z najniższymi wynikami.
Metody dokowania i projektowania śrubowego7 zastosowano w połączonym pro-2.3 w celu wygenerowania modeli konstrukcji spiralnych włókien. Poszczególne trajektorie projektowe filtrowano według następujących kryteriów: rozbieżność przekraczająca -15.0 jednostek energii Rosetty pomiędzy stanem związanym (polimerowym) i niezwiązanym (monomerycznym), powierzchnia styku przekraczająca 700 Å2, komplementarność kształtu Rosetty przekraczająca 0.62 i liczba niezadowalających reszt polarnych poniżej 5. Projekty spełniające te kryteria poddano ręcznemu udoskonalaniu, obejmującemu jednopunktowe cofnięcie się do mutacji uznanych za nie przyczyniające się do stabilizacji stanu związanego interfejsu. Projekt, który uzyskał najwyższą liczbę punktów dla każdej zadokowanej konfiguracji, został następnie zintegrowany z ostatecznym zestawem białek w celu walidacji eksperymentalnej.
Ekspresja i oczyszczanie białek
Syntetyczne geny w sumie 18 projektów zoptymalizowano pod kątem ekspresji w Escherichia coli i uzyskany z IDT, następnie wstawiony do miejsca wielokrotnego klonowania wektora pET29b+ pomiędzy miejscami restrykcyjnymi NdeI i XhoI. Konstrukty te wprowadzono do BL21* (DE3) E. coli kompetentne komórki. Transformanty hodowano w 50 ml podłoża Terrific Broth uzupełnionego 200 mg l-1 kanamycyna. Ekspresja pod kontrolą promotora T7 przebiegała przez 24 godziny w temperaturze 37°C przy użyciu autoindukcji Studiera16 do czasu zebrania kultur poprzez wirowanie. Osad komórek ponownie zawieszono w soli fizjologicznej buforowanej Tris (TBS) i poddano lizie detergentem Bugbuster. Frakcję rozpuszczalną, sklarowaną przez odwirowanie, poddano oczyszczaniu za pomocą Ni2+ chromatografia powinowactwa z unieruchomionym metalem przy użyciu żywicy Ni-NTA Superflow. Żywicę ze związanym lizatem komórkowym przemyto dziesięcioma objętościami kolumny 40 mM imidazolu i 500 mM NaCl, a następnie eluowano 400 mM imidazolem i 75 mM NaCl. Frakcje rozpuszczalne i nierozpuszczalne poddano analizie elektroforetycznej w żelu SDS-poliakryloamidowym. Do badań przesiewowych pod mikroskopem elektronowym wybrano próbki wykazujące prążki białkowe o prawidłowej masie cząsteczkowej. Wybrane projekty skalowano do 0.5 l w celu dalszej charakteryzacji, przy czym ekspresja ponownie przebiegała przez 24 godziny w temperaturze 37 °C przy użyciu autoindukcji Studiera16 przed zbiorem przez odwirowanie. Osad komórkowy ponownie zawieszono w TBS i poddano lizie metodą mikrofluidyzacji, a następnie oczyszczono jak opisano powyżej.
Barwienie negatywne EM
Rozpuszczalne frakcje zatężono w TBS (25 mM bufor Tris, 75 mM NaCl, pH 8) do badań przesiewowych pod mikroskopem elektronowym. Kroplę 6 µl (próbka 1 µl natychmiast rozcieńczona 5 µl buforu) nałożono na wyładowane ujemnie jarzeniowe, pokryte węglem miedziane siatki o oczkach 200, przemyto wodą Milli-Q i zabarwiono 0.75% mrówczanem uranylu (pH 4.0 ) lub Nano-W (pH 6.8) zakupiony od Nanoprobes, Inc., jak opisano wcześniej17. Badania przesiewowe przeprowadzono przy użyciu transmisyjnego mikroskopu elektronowego Morgagni M100 (FEI) o napięciu 268 kV lub transmisyjnego mikroskopu elektronowego Talos L120C o napięciu 120 kV (ThermoFisher). Zdjęcia wykonano za pomocą montowanej od dołu kamery Teitz CMOS 4k i przetworzono w celu zwiększenia kontrastu za pomocą oprogramowania Fiji (wersja: 2.14.0/1.54f)18 dla jasności.
Długości włókien określono ilościowo za pomocą algorytmu śledzenia włókien w CryoSPARC8. Metoda ta identyfikuje włókna poprzez korelację krzyżową z klasą szablonu i śledzenie sąsiadujących włókien ze zidentyfikowanych cząstek. Do wszystkich pomiarów włókien wykorzystano klasę szablonów wygenerowaną na podstawie DpHF19. Włókna filtrowano według średniej krzywizny (<0.0005 Å-1) i średnią znormalizowaną korelację krzyżową (> 0.5) dla każdego włókna. W przypadku DpHF18 użyliśmy 5, 2, 3, 20, 28 i 21 obrazów odpowiednio dla pH 3, 3.5, 4.2, 5, 8 i 3 do 8. W przypadku DpHF19 użyliśmy 7, 8, 8, 28, 4 i 5 obrazów odpowiednio dla pH 3, 3.5, 4.2, 5, 8 i 3 do 8. W przypadku DpHF19_9his użyliśmy 6, 6, 8, 14, 15, 8 i 4 obrazy odpowiednio dla pH 3, 3.5, 4.2, 5, 6, 8 i 3 do 8.
Cryo-EM
Próbki Cryo-EM przygotowano przez nałożenie białka na siatki dziurawo-węglowe CFLAT, odciśnięcie cieczy i zanurzenie siatek w ciekłym etanie przy użyciu Vitrobot (ThermoFisher). W przypadku DpHF19 nagrania wideo rejestrowano za pomocą mikroskopu Glacios (ThermoFisher) wyposażonego w kamerę K-2 Summit Direct Detect (Gatan Inc.) działającą w trybie zliczania, z rozmiarem piksela 1.16 Å na piksel, 50 klatek i całkowitą dawką elektronów o długości 65 Å-2. W przypadku DpHF18 i DpHF7 nagrania wideo rejestrowano za pomocą aparatu Titan Krios (ThermoFisher) wyposażonego w kamerę K-2 Summit Direct Detect (Gatan Inc.) działającą w trybie super rozdzielczości, z rozmiarem piksela 0.525 Å na piksel, 50 klatek i całkowita dawka elektronów 90 Å-2. Zautomatyzowane zbieranie danych przeprowadzono przy użyciu Leginonu19 wersja 3.4. Przetwarzanie danych przeprowadzono przy użyciu narzędzia CryoSPARC8, a przepływy pracy podsumowano na rysunkach uzupełniających. 10-12. Filmy zostały wyrównane za pomocą korekcji ruchu, a filmy o super rozdzielczości zostały podzielone na piksele o rozmiarze 1.05 Å. Parametry funkcji transferu kontrastu (CTF) oszacowano za pomocą poprawki CTF. Na podzbiorze obrazów przeprowadzono śledzenie włókien bez szablonów, a powstałe cząstki poddano klasyfikacji 2D. Wybrane klasy 2D wykorzystano następnie jako szablony do śledzenia włókien w oparciu o szablony na pełnych zbiorach danych. Po wielu rundach klasyfikacji 2D wybrane cząstki poddano udoskonaleniu 3D z narzuconą symetrią helikalną i włączoną możliwością udoskonalenia niejednorodnego. W przypadku DpHF19 narzuciliśmy symetrię helikalną z jednym początkiem, odnoszącą się do poszczególnych, niekontaktowych podjednostek, zamiast parametrów symetrii helikalnej z dwoma startami. W przypadku DpHF7 i DpHF19 udoskonalono także rozogniskowanie poszczególnych cząstek, nachylenie wiązki i aberrację sferyczną. Modyfikację gęstości przeprowadzono przy użyciu programu ResolveCryoEM w programie Phenix20,21 wersja fenix-1.20.1. Modele atomowe dla DpHF18 i DpHF19 zostały udoskonalone w mapy krio-EM przy użyciu ISOLDE22, a następnie udoskonalenie w przestrzeni rzeczywistej w programie Phenix, z wyłączonymi ograniczeniami rotamerowymi i ramachandranowymi oraz z ograniczeniami referencyjnymi narzuconymi przez wejściowy model początkowy. Do wyjaśnienia modelu dla DpHF7 wykorzystano protokół budowania modelu de novo na segmentowanej asymetrycznej gęstości jednostkowej Cryo-EM23. Późniejsze włączenie pozostałości i udoskonalenie osiągnięto przy użyciu RosettaCM24 wersja 2019.31, wykorzystująca symetrię na niesegmentowanej mapie Cryo-EM w celu uzyskania optymalnego dopasowania do gęstości i interfejsów wewnątrz włókien. Ostatnią rundę udoskonalania w przestrzeni rzeczywistej przeprowadzono w Phenixie, jak opisano powyżej dla DpHF18 i DpHF19. Statystyki gromadzenia, udoskonalania i walidacji danych Cryo-EM podsumowano w tabeli dodatkowej 1.
TIRFM
Montaż włókien
Aby zobrazować zaszczepione zarodkowanie włókien reagujących na pH, włókna DpHF18 znakowano dwoma różnymi fluoroforami sprzężonymi z maleimidem, Oregonem488 i sulfo-Cy5. Włókna znakowano 10-krotnym nadmiarem molowym, w PBS + 1 mM TCEP przez 4 godziny w temperaturze pokojowej, przed wymianą buforu na TBS (25 mM Tris, 100 mM NaCl, pH 8.0) na kolumnie wirówkowej Zeba i zatężeniu do 30 µM . Zielone włókna o stężeniu 30 µM rozmontowano poprzez dodanie 1 M cytrynianu (0.6 µl cytrynianu na 20 µl włókien) w celu obniżenia pH do 3.0. Roztwór inkubowano przez 5 minut przed dodaniem Tris (3.6 µl 1 M roztworu podstawowego), aby przywrócić pH do 8.0; Do roztworu dodano 1 µl złożonych włókien DpHF18 – Cy5 o stężeniu 30 µM. Roztwór następnie inkubowano w temperaturze pokojowej przed wirowaniem przy 13,000 XNUMX°C g przez 2 minuty w wirówce laboratoryjnej. Włókna ponownie zawieszono w TBS i obrazowano za pomocą TIRFM.
Demontaż włókien
Szybkie obrazowanie TIRFM demontażu włókien przy niskim pH wykonano na specjalnie zbudowanym systemie TIRF opartym na statywie Nikon Ti wyposażonym w system doskonałego ustawiania ostrości oraz szybki Z stolik piezoelektryczny (ASI), azymutalny oświetlacz TIRF (iLas2, Roper France) z niestandardowym rozszerzonym polem widzenia (Cairn) i obiektyw PLAN Apo 1.45 NA ×100. Obrazy uzyskano za pomocą podświetlanej od tyłu kamery sCMOS Photometrics Prime 95B, pracującej w trybie pseudoglobalnej migawki, zsynchronizowanej z oświetleniem azymutalnym. System obsługiwał Metamorph 7.10.1.161. Włókna znakowane maleimidem Sulfo-Cy5 obrazowano za pomocą lasera 630 nm (150 mW Coherent OBIS zamontowany w wyrzutni laserowej Cairn) i obrazowano za pomocą filtra Chroma ET655lp zamontowanego w kole Cairn Optospin z częstotliwością odświeżania 1 klatki na 16 ms.
Włókna obrazowano w buforze do obrazowania (25 mM Tris pH 8.0, 100 mM NaCl) w kuwecie przepływowej Ibidi zamontowanej na szkiełkach nakrywkowych do pomieszczeń czystych (niestandardowe, 25 × 75 mm2, Nexterion) i pasywowano PLL-PEG (0.1 mg/ml-1 w 20 mM Hepes, pH 7.6; 5 minut). Włókna pozostawiono do osadzenia się na szkiełku nakrywkowym przez 5 minut, po czym niezwiązane włókna usunięto za pomocą buforu obrazującego. Podczas szybkiej akwizycji pH obniżono przez przepływ w buforze o niskim pH (25 mM Tris, 100 mM NaCl, pH 3.0).
Aby zmierzyć rozkład włókien w roztworze masowym, wstępnie uformowane włókna w 1.5 ml probówkach Eppendorfa wymieniono na bufory cytrynianowe o niższym pH, aby stymulować demontaż. Część każdej reakcji pH usunięto w różnych punktach czasowych i dodano do 96-studzienkowej płytki i na 10 minut, aby umożliwić osiadanie włókien i przyleganie do szklanego podłoża. Dla każdego warunku i punktu czasowego uzyskano dziewięć pól widzenia za pomocą mikroskopu IN Cell Analyzer 2500HS (Molecular Devices) przy użyciu obiektywu powietrznego Nikon ×60 PLAN Apo 0.95 NA i źródła wzbudzenia LED o długości fali 631 nm, czas ekspozycji 150 ms z zebraną emisją przez filtr pasmowy 684 ± 24 nm. Obrazy oceniano ilościowo przy użyciu niestandardowego skryptu CellProfiler w celu segmentacji włókien za pomocą algorytmu progowania Otsu25. Dostosowywano górną i dolną granicę progu oraz okno adaptacyjne identyfikacji obiektu do momentu prawidłowej identyfikacji włókien względem sygnału tła. Długość głównej osi obiektów zidentyfikowanych za pomocą rurociągu CellProfiler wykreślono w funkcji czasu inkubacji dla każdego warunku pH.
AFM w fazie ciekłej
przygotowanie próbki
Inkubowaliśmy 10 µl 0.01% wag. roztworu polilizyny na świeżo rozszczepionej powierzchni miki muskowitowej (12 mm, Ted Pella Inc.) przez 2 minuty. Nadmiar roztworu usunięto, powierzchnię spłukano wodą i wysuszono N2 gaz7. Następnie 30 µl 10 µM roztworu białka w buforze do obrazowania (25 mM Tris-HCl, 400 mM NaCl przy pH 8) inkubowano na mice pokrytej polilizyną przez 30 minut i przemyto buforem do obrazowania w celu usunięcia nadmiaru białka. pH buforu do demontażu (25 mM Tris-HCl, 400 mM NaCl, pH 4.1, 4.4, 4, 5 lub 4.7) doprowadzono za pomocą 10 M NaOH lub 1 M kwasu cytrynowego i przed użyciem przefiltrowano przez filtr PVDF o średnicy porów 0.1 µm . Do eksperymentów z fotokwasem, 10 µM roztwór białka w 25 mM Tris-HCl pH 8 inkubowano na gołej mice przez 30 minut i przemywano 25 mM Tris-HCl pH 5.5; jeżeli gęstość liczbowa włókien na powierzchni była niska, przeprowadzano dodatkowy etap osadzania i płukania. Świeżo przygotowaliśmy również 1 mM 2-nitrobenzaldehyd (Sigma-Aldrich) w 25 mM Tris-HCl pH 5.5 i natychmiast go użyliśmy, bez ekspozycji na światło na żadnym etapie26. Pomiary spektroskopowe i pH wykazały, że 2-nitrobenzaldehyd ulega aktywacji w zakresie długości fal od 200 do 405 nm i obniża pH z 5.5 do 2.7, a wyższa intensywność lasera prowadzi do szybszego zużycia i zakwaszenia.
Obrazowanie
Do badań kinetycznych przy stałym składzie, substraty z miki polilizynowej pokryte białkiem umieszczono pod komórką płynną AFM (Bruker Multimode8). Obrazy rejestrowano w buforze obrazującym przy użyciu czystego wspornika z azotku krzemu (Bruker, SNL-10, stała sprężystości: 0.12 Nm-1, ozonowanie UV przez 5 min) w trybie pobierania w temperaturze pokojowej (25°C). Przed przepływem buforu do demontażu włókna obrazowano w sposób ciągły przez 10 minut w celu optymalizacji parametrów (256 linii skanowania, częstotliwość skanowania 1.5 Hz, wysokie wzmocnienie całkujące (3–4) i wolna amplituda 50–100 mV). Po potwierdzeniu, że nie wystąpiło żadne uszkodzenie spowodowane wspornikiem, wstrzyknięto bufor do demontażu w sposób ciągły w ilości 25 µl min-1. Konfiguracja przepływu została zoptymalizowana, aby zapewnić znikomy czas przebywania i szybkie przełączanie pH10.
Do badania fotokwasu mikę pokrytą białkiem z 25 mM Tris-HCl pH 5.5 umieszczono pod komórką płynną aparatu Cypher VRS AFM (Asylum Research) wyposażonego w laser BlueDrive (filtr intensywności x 0.3, długość fali 405 nm) z zaworem odpowietrzającym otwierane i obsługiwane w trybie dotykania. Po potwierdzeniu dużego pokrycia powierzchni włókien, bufor obrazujący zastąpiono 1 mM 2-nitrobenzaldehydem w 25 mM Tris-HCl pH 5.5, działał bez ekspozycji na widzialne światło tła i ponownie obrazowano. Następnie wspornik został cofnięty, włączono BlueDrive i wielokrotnie skanowano wstępnie wybrane obszary za pomocą zmotoryzowanego mikroskopu optycznego AFM. Całkowity czas ekspozycji na promieniowanie UV podczas rastra/zatrzymania dla wzorów punktowych i liniowych nie był dłuższy niż 10 minut, po czym wspornik przeniesiono z powrotem do naświetlonych obszarów i sfotografowano. W przypadku globalnych zmian pH okienko kwarcowe ogniwa ciekłego AFM w kontakcie z roztworem fotokwasu wystawiono na działanie ręcznej lampy UV (długość fali 364 nm) przez 7 minut, a następnie wykonano zdjęcie.
Obrazy zostały przetworzone za pomocą oprogramowania do analizy danych Gwyddion SPM v2.62 i przeanalizowane za pomocą oprogramowania Fiji v1.53s18. W przypadku kinetyki mierzono całkowitą długość włókna, a wszelkie fragmenty uznane za już zdemontowane zostały wyłączone z pomiaru długości. Aby zmierzyć szybkość demontażu na każdym końcu poszczególnych włókien (rys. 8) jako drugi koniec pomiaru długości przypisano środek włókna (połowa długości początkowej), natomiast w przypadku fragmentów włókien jako drugi koniec mierzono środek fragmentu.
- Dystrybucja treści i PR oparta na SEO. Uzyskaj wzmocnienie już dziś.
- PlatoData.Network Pionowe generatywne AI. Wzmocnij się. Dostęp tutaj.
- PlatoAiStream. Inteligencja Web3. Wiedza wzmocniona. Dostęp tutaj.
- PlatonESG. Węgiel Czysta technologia, Energia, Środowisko, Słoneczny, Gospodarowanie odpadami. Dostęp tutaj.
- Platon Zdrowie. Inteligencja w zakresie biotechnologii i badań klinicznych. Dostęp tutaj.
- Źródło: https://www.nature.com/articles/s41565-024-01641-1
- :Jest
- :nie
- ][P
- $W GÓRĘ
- 000
- 01
- 05
- 1
- 10
- 100
- 11
- 12
- 120
- 13
- 14
- 15%
- 150
- 16
- 17
- 19
- 2%
- 20
- 200
- 2001
- 2005
- 2010
- 2012
- 2013
- 2015
- 2016
- 2017
- 2018
- 2019
- 2020
- 2022
- 21
- 22
- 23
- 24
- 25
- 26
- 28
- 2D
- 30
- 31
- 35%
- 352
- 362
- 3d
- 4
- 40
- 400
- 41
- 45
- 4k
- 5
- 50
- 500
- 6
- 62
- 65
- 66
- 7
- 700
- 72
- 74
- 75
- 8
- 9
- 90
- a
- powyżej
- Stosownie
- osiągnięty
- nabyty
- nabycie
- w poprzek
- adaptive
- w dodatku
- dodatek
- Dodatkowy
- przylegać
- Skorygowana
- powinowactwo
- Po
- ponownie
- przed
- AIR
- AL
- algorytm
- Algorytmy
- wyrównany
- wyrównanie
- Wszystkie kategorie
- dopuszczać
- dozwolony
- wzdłuż
- już
- również
- an
- analiza
- Kotwica
- i
- każdy
- stosowany
- Stosowanie
- SĄ
- POWIERZCHNIA
- obszary
- AS
- zmontowane
- przydzielony
- At
- zautomatyzowane
- średni
- z dala
- Oś
- b
- z powrotem
- Kręgosłup
- tło
- na podstawie
- Belka
- zanim
- poniżej
- pomiędzy
- wiążący
- biologia
- związany
- przynieść
- bufor
- Budowanie
- objętość
- by
- aparat fotograficzny
- kandydat
- kandydatów
- Zajęte
- prowadzone
- komórka
- Komórki
- centrum
- łańcuch
- Zmiany
- wybrany
- wyjaśnione
- klarowność
- klasa
- Klasy
- klasyfikacja
- kleń
- kliknij
- ZGODNY
- gromadzone
- kolekcja
- Kolumna
- kompetentny
- w składzie
- skład
- wszechstronny
- Stężony
- stężenie
- warunek
- przeprowadzone
- systemu
- potwierdzając
- Skontaktuj się
- za
- stały
- Ograniczenia
- konstrukty
- konsumpcja
- skontaktuj się
- bez przerwy
- kontrast
- kontrola
- koordynować
- współrzędne
- Miedź
- skorygowania
- prawidłowo
- liczyć
- rachunkowość
- pokrycie
- Kryteria
- Hodowle
- zwyczaj
- Specyfikacji klienta
- szyfr
- uszkodzić
- dane
- analiza danych
- analiza danych
- Baza danych
- zbiory danych
- de
- uważane
- gęstość
- kaucja
- opisane
- Wnętrze
- zaprojektowany
- projekty
- wykryć
- determinacja
- urządzenia
- różne
- rozcieńczony
- kierować
- niepełnosprawny
- rozbieżność
- Dawka
- podczas
- e
- E i T
- każdy
- bądź
- emisja
- zatrudniony
- włączony
- zakończenia
- energia
- wzmocnione
- Środowisko
- wyposażony
- szacunkowa
- Eter (ETH)
- Każdy
- nadzwyczajny
- nadmiar
- wymiana
- wymieniony
- wyłączony
- wszechstronny
- wykazujące
- eksperymentalny
- eksperymenty
- narażony
- Ekspozycja
- wyrażenie
- dużym
- FAST
- szybciej
- fei
- pole
- Łąka
- Figa
- filtrować
- finał
- sfinalizowane
- FLOTA
- pływ
- Płynący
- Skupiać
- następnie
- następujący
- W razie zamówieenia projektu
- frakcja
- FRAME
- Francja
- Darmowy
- od
- pełny
- funkcjonować
- dalej
- Wzrost
- Generować
- wygenerowane
- szkło
- Globalne
- Zielony
- Pół
- Żniwny
- Wysoki
- wysoka rozdzielczość
- wyższy
- HTTPS
- i
- ID
- zidentyfikowane
- identyfikuje
- if
- obraz
- zdjęcia
- Obrazowanie
- natychmiast
- nałożone
- poprawa
- in
- Inc
- inkubowane
- inkubacja
- wskazany
- indywidualny
- początkowy
- wkład
- natychmiast
- integralny
- zintegrowany
- Interfejs
- interfejsy
- najnowszych
- wprowadzono
- z udziałem
- IT
- laser
- uruchomić
- Wyprowadzenia
- Doprowadziło
- Długość
- lewarowanie
- lekki
- Limity
- Linia
- linie
- LINK
- powiązany
- Ciecz
- Pętle
- niski
- niższy
- obniża
- najniższy
- poważny
- podręcznik
- mapa
- Mapy
- materiał
- Matrix
- zmierzyć
- mierzona
- pomiary
- Pomiary
- zmierzenie
- średni
- spełnione
- metal
- metoda
- metody
- Mika
- Mikroskop
- Mikroskopia
- min
- ML
- Moda
- model
- modelowanie
- modele
- Modułowa
- Cząsteczkowa
- jeszcze
- ruch
- przeniósł
- MS
- wielokrotność
- nanotechnologia
- Natura
- ujemny
- ujemnie
- Nowości
- następna generacja
- dziewięć
- Nie
- Nowy
- numer
- przedmiot
- cel
- obiekty
- miejsce
- of
- on
- na
- koncepcja
- open source
- eksploatowane
- operacyjny
- optyczny
- Optymalny
- Optymalizacja
- zoptymalizowane
- or
- na zewnątrz
- przegląd
- parametry
- Łata
- wzory
- PBS
- dla
- doskonały
- wykonywane
- Fizycznie
- rurociąg
- piksel
- umieszczony
- krok po kroku
- Platforma
- plato
- Analiza danych Platona
- PlatoDane
- zanurzenie
- punkt
- zwrotnica
- polarny
- część
- przygotowany
- premia
- PROC
- obrobiony
- przetwarzanie
- Produkcja
- Profil
- Białko
- Białka
- protokół
- zapewniać
- zakupione
- skwantyfikowany
- R
- szybki
- Kurs
- raczej
- reakcja
- Czytaj
- realistyczny
- zmniejszyć
- Zredukowany
- odniesienie
- rafinowany
- regiony
- względny
- usunąć
- Usunięto
- WIELOKROTNIE
- otrzymuje
- Badania naukowe
- Rezydencja
- Żywica
- ograniczenie
- wynikły
- sztywny
- Pokój
- okrągły
- rundy
- run
- s
- próba
- łuskowaty
- skanować
- SCI
- nauka
- wyniki
- pokaz
- scenariusz
- druga
- segment
- wybrany
- Sekwencja
- zestaw
- rozstrzygać
- Shape
- Signal
- Krzem
- pojedynczy
- witryna internetowa
- Witryny
- Rozmiar
- Miękki
- Tworzenie
- rozwiązanie
- piosenka
- Źródło
- napięcie
- specyficzność
- Spin
- Spot
- wiosna
- STAGE
- stoisko
- Startowy
- Stan
- Zjednoczone
- statystyka
- Ewolucja krok po kroku
- Stymulować
- stany magazynowe
- strategie
- Struktura
- Struktury
- Studiował
- Badanie
- kolejny
- Następnie
- Podłoże
- Szczyt
- nałożenie
- Powierzchnia
- niezrównany
- symetria
- syntetyczny
- system
- T
- stół
- talos
- dotykając
- cel
- Ted
- szablon
- Szablony
- dziesięć
- terminal
- niż
- że
- Połączenia
- Źródło
- następnie
- Te
- to
- próg
- Przez
- czas
- tytan
- do
- tolerancja
- Kwota produktów:
- Rysunek kalkowy
- przenieść
- transmisja
- Obrócony
- drugiej
- dla
- przeszedł
- jednostka
- jednostek
- aż do
- USA
- posługiwać się
- używany
- za pomocą
- v1
- uprawomocnienie
- zawór
- różnorodny
- wersja
- przez
- Filmy
- Zobacz i wysłuchaj
- widoczny
- kłęby
- W
- Wang
- była
- Uzdatnianie wody
- długości fal
- we
- waga
- DOBRZE
- były
- Koło
- natomiast
- który
- okno
- w
- bez
- przepływów pracy
- zefirnet