Materials
Solvents and reagents were purchased from commercial sources and used as received, unless otherwise specified. Methanol (ACS reagent grade) was obtained from Fisher Scientific. Molecular biology grade acrylamide (catalogue number A9099), sodium acrylate (catalogue number 408220), 19:1 acrylamide/bis-acrylamide (catalogue number A2917) and ammonium persulfate (APS; catalogue number A3678) were purchased from Sigma-Aldrich. Ultrapure N,N,N′,N′-tetramethylethylenediamine (TEMED; catalogue number 15524010) and SYBR Gold Nucleic Acid Gel Stain (catalogue number S11494) were obtained from Thermo Scientific. Desalted oligonucleotides were purchased from Integrated DNA Technologies (IDT). Nitrogen gas (>99.999%) was used under inert conditions and supplied by an in-house gas generator. To ensure an inert condition, it was purified through a Model 1000 oxygen trap from Sigma-Aldrich (catalogue number Z290246). Reagents with unreacted acrylamide groups were stored at 4 °C or −20 °C, protected from unnecessary exposure to light. Actin protein (>95% pure, rabbit skeletal muscle, catalogue number AKL95) was obtained from Cytoskeleton and DNase I (catalogue number M0303) was obtained from New England Biolabs. Matrigel was obtained from Corning (catalogue numbers 354277 and 356231). DNA ladders were purchased from Thermo Fischer (catalogue number SM1211).
Cell sources
Human bone marrow-derived MSCs were isolated from healthy female and male donors (aged 26–37) with informed consent by the Medical Faculty at the University Hospital Dresden (Ethikkommission an der Technischen Universität Dresden, ethic board number EK263122004). MDCK II cells (ECACC 00062107) were provided by A.H.’s group. Human pluripotent stem cells were generated at the CRTD Stem Cell Engineering Facility, Technische Universität Dresden, by Shahryar Khattak and registered under the hPSCreg name CRTDi003-B (https://hpscreg.eu/cell-line/CRTDi003-B). The CT27 patient-derived trophoblast stem cell (TSC) line used in this study was obtained from the RIKEN stem cell bank (https://cellbank.brc.riken.jp/cell_bank/CellInfo/?cellNo=RCB4936). The CT27 TSCs were derived from placental cytotrophoblast cells as described in ref. 62. Human placentas were obtained from healthy women with signed informed consent of the donors, and the approval of the Ethics Committee of Tohoku University School of Medicine (Research license 2014-1-879).
Polymer synthesis
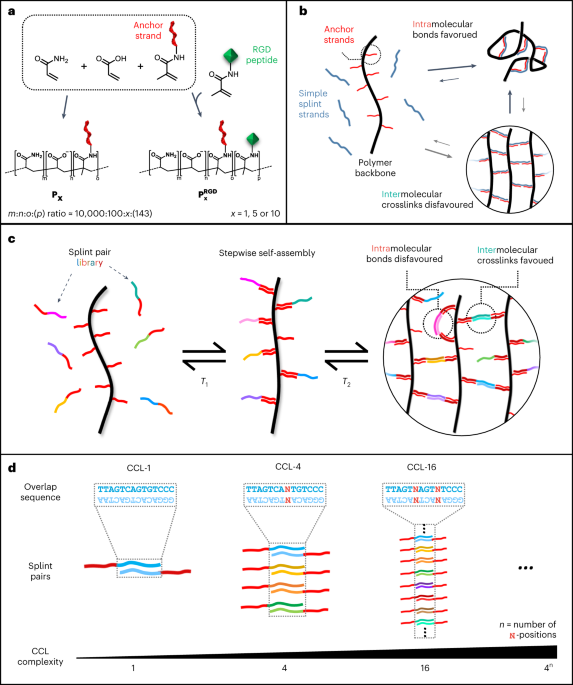
A detailed protocol is available in ref. 1) were co-polymerized in TBE buffer (100 mM Tris, 100 mM boric acid, 2 mM EDTA, pH 8.2). For P1, P5 and P10, the DNA concentrations in solution were 100 µM, 500 µM and 1,000 µM, respectively. For the corresponding RGD-functionalized derivatives ({{bf{P}}}_{{bf{5}}}^{{bf{RGD}}}) and ({{bf{P}}}_{{bf{10}}}^{{bf{R}}{bf{G}}{bf{D}}}), 10 mM of acrylated RGD peptide (sequence, G(acryl-K)GGGRGDSP) was co-polymerized with the above solution. Polymerization reactions for P1, P5 and ({{bf{P}}}_{{bf{5}}}^{{bf{RGD}}}) were initiated with 0.005 wt% APS in presence of 0.005 wt% TEMED. Synthesis of P10 and ({{bf{P}}}_{{bf{10}}}^{{bf{R}}{bf{G}}{bf{D}}}) was initiated by 0.025 wt% TEMED and 0.025 wt% APS. To achieve high molecular weight and a narrow size distribution, it was necessary to carry out the reaction in high-purity nitrogen gas, which was passed through an oxygen trap on-site. The reaction was allowed to proceed overnight, resulting in a highly viscous polymer solution indicating the formation of long polymer chains. NMR spectroscopy was used to verify high conversion of the monomer (Supplementary Fig. 3). The solution was diluted in 9 volumes of TE buffer (10 mM Tris, 1 mM EDTA, pH 8.0) and subsequently purified via methanol precipitation. The pellet was resuspended in milliQ water and stored in aliquots at −20 °C. A small aliquot of the polymer solution was lyophilized and weighted to determine the synthesis yield. Yields: 89% (P1), 98% (P5), 93% (P10), 92% (({{bf{P}}}_{{bf{5}}}^{{bf{RGD}}})) and 88% (({{bf{P}}}_{{bf{10}}}^{{bf{R}}{bf{G}}{bf{D}}})).
Nearest-neighbour thermodynamic predictions
Explicit sequences of crosslinking strands and their complements were generated using a custom Python script. The program sequentially replaces the ambiguous bases N with nucleobases (A, C, G, T) via multilayer nested loops. This created a library of forward and reverse splint variants. The minimum free energies (MFEs) of all explicit overlap domains were calculated against all explicit overlap domains in the library using NUPACK (version 4.0.0.27), using nearest-neighbour thermodynamic parameters according to ref. 63. The model parameters were set to T = 20 °C, 150 mM NaCl, 75 µM total DNA concentration (37.5 µM for each splint strand), and a complex size of 2. For each possible pair, the Boltzmann distribution factor Bx,y was sequentially calculated for each variant of forward (x) against all possible reverse splint (y) strand at the expected theoretical concentration using the equation
$${B}_{x,y}={rm{e}}^{-{{rm{MFE}}(x,y)}/{kT}}$$
where k is the Boltzmann constant. The normalized distribution was calculated to study the selective affinity of any forward splint variant in a pool of reverse splint sequences. This was calculated for each pair as the ratio of distribution factor of one pair (x, y) by the sum of all pairs as
$${B}_{x,y}^{{mathrm{normalized}}}=frac{{B}_{x,y}}{{sum }_{y}{B}_{x,y}}$$
We note that a precise solution can only be obtained by numerically solving a system of ordinary differential equations63. For simplification, we assumed that each splint strand is available at its initial concentration. In reality, non-complementary splint strand concentrations are expected to be significantly reduced, as they would be predominantly scavenged by their complementary binding partners. The obtained values for specific CCL pairs in the Boltzmann distribution that are based on our simplified calculation are therefore considered a lower limit for relative abundances.
Thermal annealing of DNA-crosslinked hydrogels
Unless specified, the samples were prepared at the final concentration of 1% (w/v) polymer solution, added with one equivalent of DNA crosslinkers to the anchor strand (for example, 75 μM crosslinkers for 1% (w/v) P5 solution), in the buffer condition of 150 mM NaCl and 1x TE buffer. The samples were annealed on the thermal cycler using the following steps: (1) heating at 95 °C for 3 min, (2) instant cooling from 95 °C to 80 °C, (3) holding at 80 °C for 2 min, (4) first cooling ramp from 80 °C to 65 °C at −0.3 °C min−1, (5) second cooling ramp from 65 °C to 37 °C at −0.5 °C min−1, (6) holding at 37 °C for 24 h.
The first cooling ramp facilitates binding of the adaptor domains of the crosslinkers to the anchor strands, while the second ramp allows proper binding of the overlap domains to their complementary partners.
Oscillatory rheological measurements
Viscoelasticity measurements were conducted using Anton Paar MCR301 rheometer with a 25-mm-diameter cone-plate geometry (cone angle 0.5°). Temperature control was ensured by installing a Peltier system PTD-200 (Anton Paar) within the measurement area. To maintain high humidity and prevent evaporation artefacts, a wet paper cylinder was placed around the plate’s circumference. Amplitude sweeps were performed from 0.1% to 1,000% strain at 1.6 Hz, while frequency sweeps were conducted from 0.1 Hz to 100 Hz at 10% strain. Temperature sweeps covered a range from 4 °C to 60 °C at 10% strain, followed by 60 °C to 70 °C at 20% strain, all at 1.6 Hz. The heating and cooling steps were performed reversibly for two cycles to exclude evaporation artefacts. Unless specified, samples were measured in triplicate for reproducibility. Stress-relaxation properties were measured at 15% strain, where the strain was kept constant while recording shear stress over time. Self-healing tests involved alternating between 1,000% and 10% strain for 5 min each, repeated 4.5 times at a frequency of 1.6 Hz. For the heat-activated gelation tests, two precursors were prepared at 1% (w/v) polymer solution, added with: (1) 40 µM forward CCL-4 strands (strand ID 4a) and 80 µM corresponding blocking strand (strand ID 14a), and (2) 40 µM reverse CCL-4 library (strand ID 4b) and 80 µM corresponding blocking strand (strand ID 14b). The precursors were heated at 95 °C for 3 min, slowly annealed with a cooling ramp from 95 °C to 4 °C at −3 °C min−1, and kept at 4 °C overnight. Before measurement, the two precursors were mixed and applied onto the rheometer. The samples were held at 4 °C for 5 min, quickly heated to 37 °C and maintained at 37 °C for 60 min with shearing at 10% strain and a frequency of 1.6 Hz.
Hydrogel printing
A microscope glass slide (76 × 26 mm, Thermo Scientific) was attached to the printing bed on BioScaffolder BS5.1 (GeSiM). Two-hundred microlitres of DyNAtrix (1% (w/v) P5 + CCL-64) stained with SYBR Gold was transferred to a disposable 1 ml syringe (Omnican 40). To remove air bubbles, the syringe was centrifuged invertedly at 300g for 2 mins. The hydrogels were printed through a 30-gauge needle (Omnican) at room temperature, with a printing speed of 2 mm s−1, an extrusion rate of 40 μm s−1 and a layer height of 0.12 mm. The resulting 3D structures, with a size of 20 × 20 × 2 mm (L × W × H), were imaged on Opera Phenix confocal microscope (PerkinElmer) at ×5 magnification.
Mixing homogeneity tests
Two precursor solutions were prepared in 1x TE buffer at the final concentration of 1% (w/v) P5 added with: (1) 40 µM forward CCL-64 strands (strand ID 6a) and 80 µM corresponding blocking strand (strand ID 15a), and (2) 40 µM reverse CCL-64 library (strand ID 6b) and 80 µM corresponding blocking strand (strand ID 15b). Ten micromolar of a 6-FAM (fluorescein)-modified strand (strand ID 15) was added to the precursor (1). After 1 h of equilibration at 4 °C, the two precursors were mixed using a positive displacement pipette. The mixture was dispensed into a 384-well plate (Greiner Bio-One) at a volume of 10 µl per well. The plate was then incubated at 37 °C for 30 min to initiate gelation. Finally, images were acquired on Andor Dragonfly Confocal Microscope (Oxford Instrument) at ×4 magnification.
Enzymatic digestion assay
The Förster-resonance-energy-transfer-paired oligos, modified with Cy5 fluorophore and Iowa Black Dark quencher (Q), were purchased from IDT. A mixture of 2 µM Cy5-strand (strand ID 13a) and 4 µM Q-strand (strand ID 13b) was prepared in 1x PBS buffer and subjected to annealing steps: heating at 95 °C for 1 min, instant cooling to 50 °C for 2 min and gradual cooling from 50 °C to 20 °C at a rate of −1.5 °C min−1. Samples were prepared with a final concentration of 100 nM Cy5-Q Förster-resonance-energy-transfer-probe and varying actin content from 2.5 µg ml−1 to 320 µg ml−1 (Cytoskeleton, catalogue number AKL95-B) in a buffer containing 150 mM NaCl, 1.8 mM CaCl2, 0.2 mM ATP (Jena Bioscience, catalogue number NU-1010) and 1x IDTE. Before measurement, 80 U ml−1 DNase I (NEB, catalogue number M0303S) was added to each sample. Each sample was loaded in triplicate at a volume of 20 µl per well into a 96-well quantitative PCR plate. Fluorescence signals were recorded every 10 min for 24 h, and subsequently every 30 min for the following 36 h at 37 °C using a Bio-Rad CFX96 Real-Time PCR System.
Gel volume and digestion measurements
The lyophilized actin protein (Cytoskeleton, catalogue number AKL95-B) was reconstituted with 100 µl deionized water to a concentration of 10 mg ml−1. The reconstituted actin was then in a buffer containing 5 mM Tris-HCl pH 8.0, 0.2 mM CaCl2, 0.2 mM ATP, 5% (w/v) sucrose and 1% (w/v) dextran. Citric acid powder (192.124 MW) was prepared in 1x PBS to a final concentration of 200 mM, pH 7.0. The gel (1% (w/v) P5 crosslinked with CCL-64) were stained with 10X SYBR Gold before swelling. The gel was then dispensed into a 384-well plate (Greiner, number 788092) at a volume of 5 µl per well. The samples were immersed in 20 µl of (1) 1x TE buffer, (2) 10% FBS-containing DMEM, (3) 50 µg ml−1 actin + 10% FBS-containing DMEM, or (4) 10 mM citrate + 10% FBS-containing DMEM. Images were captured every hour for 48 h at 37 °C on Opera Phenix Plus confocal microscope (PerkinElmer) at ×5 magnification. The gel volume was quantified using IMARIS software (Oxford Instrument) through the surface wizard and statistical tool under a uniform threshold setting.
Whole blood incubation
Whole blood incubation was performed as described previously64,65. Gels were prepared on glass carriers with a polyethylene-alt-maleic anhydride bonding layer66, and inserted in in-house developed incubation chambers made of polytetrafluoroethylene, where a 3.2 cm2 test surface was exposed to 2 ml blood64. Reactively cleaned glass67 and Teflon AF (DuPont) served as activating and inert reference surfaces, respectively.
Blood was obtained from two voluntary ABO-matched donors who had not used any medicine in the past ten days. The blood was immediately anticoagulated with 1.0 U ml−1 heparin (Ratiopharm), pooled and filled into the incubation chambers with the samples, avoiding an air interface. The chambers were incubated for 2 h at 37 °C at constant overhead rotation to prevent sedimentation. The blood was subsequently analysed for the blood cell count (Beckmann Coulter AcTdiff). Granulocyte and monocyte activation were determined by flow cytometry (LSR Fortessa, Becton Dickinson), where granulocytes and monocytes were identified for their scatter characteristics and CD15 (clone SSEA-1, PE-conjugated, BioLegend) and CD14 (clone M5E2, APC conjugated, Becton Dickinson) positivity, respectively. The intensity of CD11b expression (clone ICRF44, PacificBlue-conjugated, BioLegend) was normalized to blood incubated with 100 EU ml−1 endotoxin. The rate of granulocytes with conformationally activated CD11b was determined using anti-CD11b clone CBRM1/5 (PE/Cy7-conjugated, BioLegend).
Soluble markers of coagulation activation (prothrombin fragment F1+2), blood platelet activation (platelet factor 4 (PF4)) and complement activation (complement fragment C5a) were determined from plasma using commercial enzyme-linked immunosorbent assays (ELISAs ; Enzygnost F1+2, Siemens Healthineers; Zymutest PF4, Hyphen BioMed; C5a ELISA, DRG Instruments) after stabilization of the blood with the recommended additives of the test kits and centrifugation.
The analysis was performed with a triplicate set of samples in parallel (n = 3). The study was covered by the ethic vote EK-BR-24/18-1 of the Sächsische Landesärztekammer.
3D cell culture
A detailed protocol is available in Supplementary Methods. In brief, gel components were dissolved in water and stored at −20 °C for cell culture. For MSC and hiPSC cultures, two precursors were prepared at a final concentration of 1% (w/v) ({{bf{P}}}_{{bf{5}}}^{{bf{RGD}}}) with: (1) 37.5 µM forward splint (strand ID 6a) and 75 µM blocking strand (strand ID 15a), and (2) 37.5 µM reverse splint strands (strand ID 6b) and 75 µM blocking strand (strand ID 15b). As for MDCKII and hTSC cultures, two precursors were prepared at a final concentration of 1% (w/v) ({{bf{P}}}_{{bf{10}}}^{{bf{RGD}}}) with: (1) 75 µM forward splint (strand ID 6a) and 150 µM blocking strand (strand ID 15a), and (2) 75 µM reverse splint strands (strand ID 6b) and 150 µM blocking strand (strand ID 15b). Concentrated DMEM was added to reach a final 1x concentration. The precursors were equilibrated at 4 °C overnight for the blocking strands to bind quantitatively to the splint strands.
The cell suspension was mixed with the first precursor (1) and then with the second precursor (2). Cell-laden hydrogels (5 µl per well) were placed in 384-well plates or micro-well inserts (ibidi, catalogue number 80409) attached in 6-well plates. The samples were incubated at 37 °C for 30 min to trigger heat-activated gelation. After gelation, individual culture medium was added to each well. When using serum-containing medium, 100 µg ml−1 of actin (Cytoskeleton, catalogue number AKL95) was included to suppress nuclease activity. The embedded cells were cultured at 37 °C, 5% CO2.
For Matrigel groups, cells were embedded in 50% Matrigel. The cell seeding density, gelation time and medium were identical to the DyNAtrix samples.
Fluorescence live/dead assay and cell release
MSCs were stained with 3 µM calcein AM (PromoKine, catalogue number PK-CA707-80011) and 0.75 µM Draq7 (Invitrogen, catalogue number D15106) in medium for 30 min at 37 °C, 5% CO2. Confocal images were taken on Opera Phenix Plus confocal microscope (PerkinElmer) at ×10 magnification. Cell counting was performed with IMARIS software (Oxford Instrument) using the spots wizard. Cell viability was determined by dividing the number of live cells by the sum of live and dead cells.
For cell release, 2 U per well DNase I was added after the MSCs were stained with 3 µM calcein AM for 30 min. Confocal images were acquired every 20 min at ×5 magnification for 2 h at 37 °C with 5% CO2.
Immunofluorescent staining
hiPSCs were fixed with 2% paraformaldehyde for 40 min at room temperature, followed by permeabilization and blocking with 0.1% Triton X-100 in 2% BSA/PBS for 1 h °C. The cells were then incubated overnight at 4 °C with the primary antibodies (1:300 anti-Oct3/4 (BD Biosciences, catalogue number 611202)) in 2% BSA/PBS. After washing the samples three times with PBS, secondary antibodies (1:200 Alexa Fluor 488, 1:200 Phalloidin ATTO 550 and 1:1,000 Hoechst 33342) in 2% BSA/PBS were added and incubated overnight at 4 °C. Confocal images were acquired using Opera Phenix Plus confocal microscope (PerkinElmer).
As for hTSCs and trophoblast organoids, the samples were fixed with 2% paraformaldehyde for 40 min at 4 °C, permeabilized with 0.5% Tween-20/PBS for 30 min and blocked with 3% BSA (Sigma) + 0.1% Tween-20 for 1 h. The cells were incubated with the primary antibodies in the blocking solution for 2 days at 4 °C. The primary antibodies used were: anti-E-cadherin (Invitrogen, catalogue number 13-1900, 1:200), anti-Syndecan (Sigma, catalogue number HPA006185, 1:200), anti-GATA3 (R&D, catalogue number AF2605, 1:200), anti-ENDOU (Sigma, catalogue number HPA012388, 1:200), anti-GCM1 (Atlas, catalogue number HPA011343, 1:200) and anti-TEAD4 (abcam, catalogue number ab58310, 1:200). After washing with PBS 3 times, the cells were then incubated with the secondary antibodies in the blocking solution for 2 days at 4 °C. The secondary antibodies used were: Alexa Fluor 488-, 594- and 647-conjugated antibodies. Nuclei were stained with Hoechst (1:200). Confocal images were acquired using Zeiss LSM 880 Airy inverted microscope and the ×63/1.3 LCI Plan-Neofluar objective with water immersion medium.
Ethics statement
Informed consent was obtained from all recipients and/or donors of cells or tissues. The study involving human whole blood was covered by the ethic vote EK-BR-24/18-1 of the Sächsische Landesärztekammer. The blood was obtained from two voluntary ABO-matched donors who had not used any medicine in the past ten days. The study involving human bone marrow-derived MSCs was covered by the ethic vote EK221102004 and EK47022007 at TU Dresden. MSCs were isolated from healthy female and male donors (aged 26–37) by the Medical Faculty at the University Hospital Dresden. The study involving hiPSCs was covered by the ethic vote EK363112012 at TU Dresden. hiPSCs were generated from MACS-sorted CD34+ cells from the peripheral blood of a healthy donor (aged 20–24). The CT27 patient-derived trophoblast stem cell line used in this study had been derived from placental cytotrophoblast cells and was obtained from the RIKEN stem cell bank (RCB4936:CT27). Human placentas were obtained from healthy women with signed informed consent of the donors, and the approval of the Ethics Committee of Tohoku University School of Medicine (research license 2014-1-879).
Statistics and reproducibility
Graphs and statistical analyses were made in GraphPad Prism and Origin 2022. Sample sizes are specified in the figure captions, where n refers to the number of distinct samples. For the cell viability assay, a one-way analysis of variance (ANOVA) test with a Tukey post hoc test was performed (n = 3, df = 4). For the blood compatibility tests, a one-way ANOVA with a Holm–Sidak post hoc test was performed (n = 3, df = 6). Data are expressed as mean ± s.d., with *P < = 0.05, **P < = 0.01 and ***P < = 0.001 indicating statistical significance. No statistical method was used to determine sample size, and no data were excluded from the analyses. The experiments were not randomized, and the investigators were not blinded to allocation during experiments and outcome assessment.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
- SEO Powered Content & PR Distribution. Get Amplified Today.
- PlatoData.Network Vertical Generative Ai. Empower Yourself. Access Here.
- PlatoAiStream. Web3 Intelligence. Knowledge Amplified. Access Here.
- PlatoESG. Automotive / EVs, Carbon, CleanTech, Energy, Environment, Solar, Waste Management. Access Here.
- BlockOffsets. Modernizing Environmental Offset Ownership. Access Here.
- Source: https://www.nature.com/articles/s41565-023-01483-3
- :is
- :not
- :where
- ][p
- 000
- 1
- 10
- 100
- 12
- 14
- 15%
- 19
- 1998
- 1999
- 2%
- 20
- 200
- 2012
- 2018
- 2020
- 2022
- 22
- 24
- 26
- 27
- 30
- 300
- 320
- 33
- 36
- 3d
- 40
- 50
- 500
- 60
- 65
- 66
- 67
- 7
- 70
- 75
- 8
- 80
- 9
- 95%
- a
- above
- According
- Achieve
- acquired
- activated
- activating
- Activation
- activity
- added
- additives
- After
- against
- aged
- AIR
- AL
- Alexa
- All
- allocation
- allowed
- allows
- am
- an
- analysis
- Anchor
- and
- Antibodies
- any
- applied
- approval
- ARE
- AREA
- around
- article
- AS
- assessment
- assumed
- At
- atlas
- attached
- available
- avoiding
- b
- Bank
- based
- BD
- BE
- been
- before
- between
- bind
- binding
- biology
- Biomaterials
- Black
- blocked
- blocking
- blood
- board
- BONE
- BSA
- buffer
- by
- calculated
- CAN
- captions
- captured
- carriers
- carry
- cell
- Cells
- chains
- characteristics
- chemical
- Cleaning
- click
- commercial
- committee
- comparison
- compatibility
- Complement
- complementary
- complex
- components
- Concentrated
- concentration
- condition
- conditions
- conducted
- consent
- considered
- constant
- content
- control
- Conversion
- Corresponding
- counting
- covered
- created
- Culture
- custom
- cycles
- Dark
- data
- Days
- dead
- density
- Derivatives
- Derived
- described
- Design
- detailed
- Determine
- determined
- developed
- distinct
- distribution
- dna
- domains
- donors
- Dragonfly
- during
- dynamic
- e
- E&T
- each
- embedded
- Engineering
- England
- ensure
- Equivalent
- Ether (ETH)
- Ethic
- ethics
- EU
- evaluation
- Every
- example
- excluded
- expected
- experiments
- exposed
- Exposure
- expressed
- expression
- facilitates
- Facility
- factor
- female
- Fig
- Figure
- filled
- films
- final
- Finally
- First
- fixed
- flow
- followed
- following
- For
- formation
- Forward
- Free
- Frequency
- from
- GAS
- generated
- generator
- geometry
- glass
- Gold
- grade
- gradual
- Group
- Group’s
- had
- healthy
- height
- Held
- High
- highly
- holding
- Hospital
- hour
- HTTPS
- human
- i
- ID
- identical
- identified
- ii
- images
- immediately
- immersed
- immersion
- Impact
- in
- included
- incubated
- INCUBATION
- indicating
- individual
- information
- informed
- initial
- initiate
- initiated
- Inserts
- installing
- instant
- instrument
- instruments
- integrated
- Interface
- into
- Investigators
- involved
- involving
- Iowa
- isolated
- IT
- ITS
- kept
- layer
- Library
- License
- light
- LIMIT
- Line
- LINK
- linked
- live
- Long
- lower
- made
- maintain
- material
- mean
- measured
- measurement
- measurements
- medical
- medicine
- medium
- Methanol
- method
- methods
- Microscope
- min
- minimum
- mixed
- mixture
- ML
- model
- modified
- molecular
- name
- nanotechnology
- narrow
- Nature
- necessary
- New
- no
- note
- novel
- number
- numbers
- objective
- obtained
- of
- on
- ONE
- only
- onto
- Opera
- or
- ordinary
- Origin
- otherwise
- our
- out
- Outcome
- over
- overnight
- Oxford
- Oxygen
- pair
- pairs
- Paper
- Parallel
- parameters
- partners
- passed
- past
- PBS
- PCR
- per
- performed
- peripheral
- Plasma
- platform
- plato
- Plato Data Intelligence
- PlatoData
- plus
- polymer
- pool
- portfolio
- positive
- Positivity
- possible
- Post
- precise
- precursor
- predominantly
- preparation
- prepared
- presence
- prevent
- primary
- printing
- PROC
- processing
- Program
- proper
- properties
- protected
- Protein
- protocol
- provided
- purchased
- Python
- quantitative
- quickly
- R
- R&D
- Rabbit
- Ramp
- Randomized
- range
- Rate
- ratio
- reach
- reaction
- reactions
- real-time
- Reality
- received
- recipients
- recommended
- recorded
- recording
- Reduced
- reference
- refers
- registered
- relative
- release
- remove
- repeated
- Reporting
- research
- resulting
- reverse
- RIKEN
- Room
- s
- School
- SCI
- scientific
- Second
- secondary
- selective
- Sequence
- set
- setting
- Siemens
- Sigma
- signals
- signed
- significance
- significantly
- silk
- simplified
- Size
- sizes
- Slide
- Slowly
- small
- smart
- sodium
- Software
- solid
- solution
- Solutions
- Solving
- Sources
- specific
- specified
- Spectroscopy
- speed
- spots
- statistical
- Stem
- stem cells
- Steps
- stored
- Strands
- stress
- Study
- Subsequently
- supplied
- Surface
- suspension
- system
- Systems
- T
- table
- taken
- targets
- Technologies
- ten
- test
- tests
- that
- The
- their
- then
- theoretical
- therefore
- thermal
- they
- this
- three
- threshold
- Through
- time
- times
- tissues
- to
- tool
- Total
- transferred
- trigger
- Triton
- two
- under
- unified
- university
- unnecessary
- USA
- used
- using
- Values
- Variant
- verify
- versatile
- version
- via
- viability
- View
- volume
- volumes
- voluntary
- Vote
- was
- washing
- Water
- we
- weight
- WELL
- were
- when
- which
- while
- WHO
- whole
- with
- within
- Women
- would
- X
- Yield
- yields
- zephyrnet